Organolitio
Un reactivo de organolitio es un compuesto organometálico con un enlace covalente entre un átomo de carbono y uno de litio. Como la naturaleza electropositiva del litio hace que la densidad de carga del enlace esté sobre el átomo de carbono, se crea efectivamente un carbanión. De ahí que los compuestos de organolitio sean bases y nucleófilos extremadamente potentes.
Preparación
editarLa mayoría de los reactivos de alquil-litio simples y las amidas de litio comunes están disponibles comercialmente en una variedad de disolventes y concentraciones. Los reactivos de organolitio también se pueden preparar en el laboratorio. A continuación se presentan algunos métodos comunes para preparar reactivos de organolitio.
Reacción con litio metal
editarLos reactivos de organolitio se preparan industrialmente por la reacción de un halocarbono con litio metálico, esto es:[1][2]
|
|
(1) |

La preparación industrial de reactivos de organolitio se logra utilizando este método, al tratar el cloruro de alquilo del organolitiado que se quiere obtener con litio metálico y un 0,5 a 2% de sodio. La conversión es altamente exotérmica. El sodio inicia la vía radical y aumenta la velocidad.[3] La reducción procede a través de una vía radical. A continuación se muestra un ejemplo de la preparación de un reactivo de litio funcionalizado mediante reducción con metal de litio.[4] A veces, el metal de litio en forma de polvo fino se usa en la reacción con ciertos catalizadores como el naftaleno o el 4,4'-di-t-butilbifenilo (DTBB). Otro sustrato que se puede reducir con litio metálico para generar reactivos de alquil-litio son los sulfuros. La reducción de sulfuros es útil en la formación de reactivos de organolitio funcionalizados tales como alfa-litio éteres, sulfuros y silanos.[5]

Reduction with Li metal

En lugar de litio elemental, también se pueden usar compuestos de organolitio disponibles comercialmente como reactivos de litiación. Estos sufren una reacción de metátesis con organohaluros para formar el compuesto litiado y el haluro de alquilo correspondiente:
- Litiación de un areno, Ar = Aril, X = Halogenuro.

Metalación
editarUn segundo método para preparar reactivos de organolitio es una metalación (intercambio de litio e hidrógeno). La acidez relativa de los átomos de hidrógeno controla la posición de litiación. La desprotonación de la sustancia a litiar con compuestos de organolitio disponibles comercialmente, como el n-butil-litio, donde el orgolitio extrae un protón activado, como resultado de lo cual se forma el organolitio deseado y se libera el alcano en el que se basa el organolitio utilizado (en el ejemplo butano):

Sin embargo, esta reacción requiere que el organolitio formado sea una base más débil que el organolitio original. Dado que el organolitio utilizado es fuertemente básico y, en consecuencia, reacciona con baja selectividad, el protón a extraer debe ser significativamente más ácido que otros protones presentes en la molécula a litiar.
Este es el método más común para preparar reactivos de alquinil-litio, porque el hidrógeno terminal unido al carbono sp es muy ácido y se desprotona fácilmente.[2]
Para los compuestos aromáticos, la posición de litiación también está determinada por el efecto de dirección de los grupos sustituyentes.[6] Algunos de los grupos sustituyentes directores más efectivos son alcoxi, amido, sulfóxido, sulfonilo. La metalación a menudo ocurre en la posición orto a estos sustituyentes, ya que estos tienen pares de electrones solitarios que estabilizan el ion de litio y, por lo tanto, lo acercan espacialmente al protón en posición orto , que luego puede abstraerse preferentemente.

En los compuestos heteroaromáticos, la metalación generalmente ocurre en la posición orto respecto al heteroátomo.[2][6]
En la siguiente imagen se resumen algunos de los compuestos de organolitio que se pueden obtener por metalación:

Intercambio halógeno-litio
editarUn tercer método para preparar reactivos de organolitio es el intercambio de litio-halógeno, que implica el intercambio de heteroátomos entre un organohaluro y una especie de organolitio.
|
|
(2) |

El intercambio de litio-halógeno es muy útil en la preparación de nuevos reactivos de organolitio.
El terc-butil-litio o el n-butil-litio son los reactivos más utilizados para generar nuevas especies de organolitio a través del intercambio de halógeno de litio. El intercambio de litio-halógeno se usa principalmente para convertir yoduros y bromuros de arilo y alquenilo con carbonos sp2 en los compuestos de organolitio correspondientes. La reacción es extremadamente rápida y, a menudo, transcurre entre -60 y -120 °C.[7]
Transmetalación
editarLos reactivos de organolitio se pueden preparar a partir de otros compuestos organometálicos por transmetalación. Los compuestos de organocobre, organoestaño, organosilicio, organoboro, etc.
|
|
(3) |

Los tipos comunes de transmetalación incluyen el intercambio Li/Sn, Li/Hg y Li/Te, que son rápidos a baja temperatura.[8] La ventaja del intercambio de Li/Sn es que los precursores de trialquilestannano experimentan pocas reacciones secundarias, ya que los subproductos de n-Bu3Sn resultantes no reaccionan frente a los reactivos de alquil-litio.[8] En el siguiente ejemplo, el vinilestannano, obtenido por hidroestannilación de un alquino terminal, forma vinil-litio a través de la transmetalación con n-BuLi.[9]
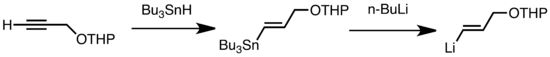
Li Sn exchange

Estructura
editarAunque las especies de alquil-litio simples a menudo se representan como monómero RLi, existen como agregados (oligómeros) o polímeros.[10] El grado de agregación depende del sustituyente orgánico y de la presencia de otros ligandos, de forma que el litio se coordine con más de un átomo de carbono, y cada átomo de carbono se coordine con más de un átomo de litio. Tres factores generales afectan la agregación: la interacción electrostática entre cargas opuestas, la esfera de coordinación del litio puede incluir moléculas de disolvente o base de Lewis, y el impedimento estérico de la parte hidrocarbonada.[11].[12] Los átomos de litio tienden a formar triángulos y agregados superiores.Estas estructuras se han dilucidado mediante una variedad de métodos, en particular espectroscopía de RMN de6Li, 7Li, y 13C y análisis de difracción de rayos X.[13] La química computacional respalda estas asignaciones.[10]
Naturaleza del enlace carbono-litio
editar
Las electronegatividades relativas del carbono y el litio sugieren que el enlace C−Li será altamente polar.[14][15][16] Sin embargo, ciertos compuestos de organolitio poseen propiedades como la solubilidad en solventes no polares que complican el problema.[14] Si bien la mayoría de los datos sugieren que el enlace C-Li es esencialmente iónico, se ha debatido si existe un pequeño carácter covalente en el enlace C-Li.[15][16] Una estimación sitúa el porcentaje de carácter iónico de los compuestos de alquillitio entre el 80 y el 88 %.[17]
En los compuestos de alil-litio, el catión de litio se coordina con la cara del enlace de carbono π en una forma η3 en lugar de un centro carbaniónico localizado, por lo tanto, los alil-litio suelen estar menos agregados que los alquil-litio.[12][18] En los complejos de aril-litio, el catión de litio se coordina con un solo centro carbaniónico a través de un enlace de tipo Li−C σ.[12][19]



Estructura en estado sólido
editar
Al igual que otras especies que consisten en subunidades polares, las especies de organolitio se agregan.[20][21] La formación de agregados está influenciada por las interacciones electrostáticas, la coordinación entre el litio y las moléculas de disolvente circundantes o los aditivos polares y los efectos estéricos.[10]
Un componente básico para la construcción de estructuras más complejas es un centro carbaniónico que interactúa con un triángulo Li3 en forma de η-3. En los reactivos de alquil-litio simples, estos triángulos se agregan para formar estructuras de tetraedro u octaedro.
Por ejemplo, en el metillitio, etil-litio y terc-butil-litio en estado sólido , 4 átomos de litio forman un tetraedro con cada cara tapada por un grupo metilo, etilo o terc-butilo [RLi]4 que enlaza simultáneamente con 3 átomos de litio (η3 hapticidad).[10] El metil-litio existe como tetrámeros en un grupo de tipo cubano en estado sólido, con cuatro centros de litio que forman un tetraedro. Cada metanuro en el tetrámero del metil-litio puede tener una interacción agóstica con los cationes de litio en los tetrámeros adyacentes.[10][20] El etillitio y el terc-butil-litio, por otro lado, no muestran esta interacción y, por lo tanto, son solubles en disolventes de hidrocarburos no polares. Otra clase de alquil-litio adopta estructuras hexámeras, como n-butil-litio, isopropil-litio y ciclohexanil-litio.[10]
La adición de una base de Lewis como los disolventes dietiléter o THF, o los ligandos nitrogenados TMEDA, PMDTA o esparteina tiende a desagregar los compuestos de organolitio, haciéndolos más solubles y reactivos. El complejo de estado sólido de MeLi con (-)-esparteina es un dímero. El complejo de butillitio con PMDTA está muy próxino al monómero BuLi.
Estructura en disolución
editarConfiar únicamente en la información estructural de los agregados de organolitio obtenidos en estado sólido a partir de estructuras cristalinas tiene ciertos límites, ya que es posible que los reactivos de organolitio adopten diferentes estructuras en el entorno de la solución de reacción.[12] Además, en algunos casos, la estructura cristalina de una especie de organolitio puede ser difícil de aislar. Por lo tanto, estudiar las estructuras de los reactivos de organolitio y los intermedios que contienen litio en forma de disolución es extremadamente útil para comprender la reactividad de estos reactivos.[22] La espectroscopia de RMN se ha convertido en una poderosa herramienta para el estudio de agregados de organolitio en disolución.
En disolución, el metil-litio en THF a concentración 1M es un tetrámero, el n-butil-litio en benceno a concentración 3M es un hexámero y en THF a concentración 1M es un tetrámero. El t-BuLi en THF es un dímero. El isopropil-litio en ciclopentano es una mezcla de las formas hexámero, octámero y nonámero.
Diferentes estados de agregación de organolitio se encuentran en la desprotonación simple del alquino terminal (feniltio)acetileno por n-butillitio en THF a -135 °C, un proceso que puede ser seguido por espectroscopia RMN de 7Li:[23]

El tetrámero de una sustancia parecida al cubano A es fuertemente reactivo comparado con eldímero B que forma en primer lugar laespecie de mezcla de dímeros C y finalmente el homodímero D. De hecho, el dímero es más reactivo que el tetrámero en un factor de 3.2x108.
| Disolvente | Estructura | |
|---|---|---|
| metil-litio | Hidrocarburo | hexámero (octaedro Li6) |
| metil-litio | THF, éter, HMPA | tetrámero (tetraedro Li4) |
| metil-litio | TMEDA | monómero |
| n-butil-litio | pentano, ciclohexano | hexámero |
| n-butil-litio | éter | tetrámero |
| n-butil-litio | THF | tetrámero-dímero |
| sec-butil-litio | pentano | hexámero-tetrámero |
| isopropil-litio | pentano | hexámero-tetrámero |
| terc-butil-litio | pentano | tetrámero |
| terc-butil-litio | THF | monómero |
| fenil-litio | éter,THF, HMPA | dímero |
| bencil-litio | THF, éter | monómero |
Los reactivos de organolitio también reaccionan con los disolventes tipo éter que son muy usados en la mayoría de las reacciones. La tabla inferior muestra la vida media aproximada de varios compuestos de organilitio en disolventes típicos.:[25]
| Disolvente/ Temperatura |
n-butillitio ( n-BuLi ) |
s-butillitio ( s-BuLi ) |
t-butillitio ( t-BuLi ) |
metillitio (MeLi ) |
CH2=C(OEt)-Li | CH2=C(SiMe3)-Li |
|---|---|---|---|---|---|---|
| THF/-20 °C | 40 min, 360 min | |||||
| THF/20 °C | >15 h | 17 h | ||||
| THF/35 °C | 10 min | |||||
| THF/TMEDA/-20 °C | 3300 min | |||||
| THF/TMEDA/ 0 °C | 340 min | |||||
| THF/TMEDA/20 °C | 40 min | |||||
| Éter/-20 °C | 480 min | |||||
| Éter/0 °C | 61 min | |||||
| Éter/20 °C | 153 h | <30 min | 17 days | |||
| Éter/35 °C | 31 h | |||||
| Éter/TMEDA/ 20 °C | 603 min | |||||
| DME/-70 °C | 120 min | 11 min | ||||
| DME/-20 °C | 110 min | 2 min | <<2 min | |||
| DME/0 °C | 6 min |
Reactividad y aplicaciones
editarComo ya se ha comentado, el enlace C-Li en los reactivos de organolitio están fuertemente polarizados debido al carácter electropositivo del litio. Como resultado, el carbono atrae la mayor parte de la densidad electrónica del enlace y se parece a un carbanión. Son, por lo tanto, fuertemente básicos y nucleófilos altamente reactivos y reaccionan con casi todos los tipos de electrófilos. Son semejantes a los reactivos de Grignard, pero son mucho más reactivos. Debido a esta reactividad, son incompatibles con agua, oxígeno (O2), y dióxido de carbono, y deben ser manejados bajo atmósfera protectora como nitrógeno o, preferiblemente, argón. Algunas de las aplicaciones más comunes de los reactivos de organolitio en síntesis incluyen su uso como nucleófilos, bases fuertes para la desprotonación, iniciador de la polimerización y material de partida para la preparación de otros compuestos organometálicos.
Uso como nucleófilo
editarReacciones de carbolitiación
editarComo nucleófilos, los reactivos de organolitio se someten a reacciones de carbolitiación, en las que el enlace carbono-litio se adiciona a un enlace múltiple de carbono-carbono (adición a un doble enlace o triple enlace), formando nuevas especies de organolitio.[26] Esta reacción es la reacción más ampliamente empleada de compuestos de organolitio. La carbolitiación es clave en los procesos de polimerización aniónica, y el n-butillitio se usa como catalizador para iniciar la polimerización de estireno, butadieno o isopreno o sus mezclas.[27][28]

Anionic polymerization of styrene initiated by sec-butyllithium

Otra aplicación que aprovecha esta reactividad es la formación de compuestos carbocíclicos y heterocíclicos por carbolitiación intramolecular.[26] Como forma de ciclación aniónica, las reacciones de carbolitiación intramolecular ofrecen varias ventajas sobre la ciclación por radicales. En primer lugar, es posible que las especies de organolitio cíclicas del producto reaccionen con electrófilos, mientras que a menudo es difícil atrapar un radical intermedio de la estructura correspondiente. En segundo lugar, las ciclaciones aniónicas suelen ser más regio y estereoespecíficas que las ciclaciones por radicales, particularmente en el caso de los 5-hexenillitios. La carbolitiación intramolecular permite la adición de alquil-vinil-litio a enlaces triples y dobles enlaces sustituidos con mono-alquilo. Los arillitios también pueden sufrir una adición si se forma un anillo de 5 miembros. Las limitaciones de la carbolitiación intramolecular incluyen la dificultad de formar anillos de 3 o 4 miembros, ya que las especies de organolitio cíclicas intermedias a menudo tienden a sufrir aperturas de anillo.[26] A continuación se muestra un ejemplo de reacción de carbolitiación intramolecular. Las especies de litio derivadas del intercambio litio-halógeno se ciclaron para formar el vinil-litio a través del cierre del anillo 5-exo-trig. La especie de vinil-litio reacciona además con electrófilos y produce compuestos de ciclopentilideno funcionalizados.[29]

A sample stereoselective intramolecular carbolithiation reaction

Adición a compuestos carbonílicos
editarLos compuestos de organolitio se usan también habitualmente para reacciones de adición nucleofílica a compuestos con el grupo carbonilo (C=O) para dar alcoholes y otros átomos de carbono electrófilos. La desprotonación puede ser una reacción colateral con compuestos de carbonilo enolizable,especialmente con reactivos de organolitio con impedimento estérico tales como t-butil-litio. Los reactivos de Grignard, aunque mucho menos reactivos, son una alternativa en reacciones de adición, con menos problemas con la desprotonación. La adición procede principalmente a través de la adición polar, en la que las especies de organolitio nucleofílicas atacan desde la dirección ecuatorial y producen el alcohol axial.[2] La adición de sales de litio como LiClO4 puede mejorar la estereoselectividad de la reacción.[30]

LiClO4 increase selectivity of t BuLi

Cuando la cetona está estéricamente impedida, el uso de reactivos de Grignard a menudo conduce a la reducción del grupo carbonilo en lugar de la adición.[2] Sin embargo, es menos probable que los reactivos de alquillitio reduzcan la cetona y se pueden usar para sintetizar alcoholes sustituidos.[31] A continuación se muestra un ejemplo de adición de etillitio a adamantona para producir alcohol terciario.[32]
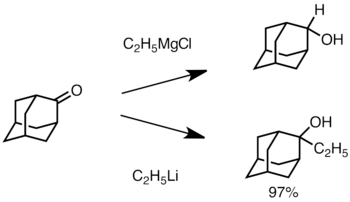
Li add to adamantone

Los reactivos de organolitio también son mejores que los reactivos de Grignard en su capacidad para reaccionar con ácidos carboxílicos para formar cetonas.[2] Esta reacción se puede optimizar controlando cuidadosamente la cantidad de reactivo de organolitio que se agrega o usando cloruro de trimetilsililo para apagar el exceso de reactivo de litio.[33] Una forma más común de sintetizar cetonas es mediante la adición de reactivos de organolitio a las amidas de Weinreb (N-metoxi-N-metilamidas). Esta reacción proporciona cetonas cuando los reactivos de organolitio se usan en exceso, debido a la quelación del ion de litio entre el oxígeno N-metoxi y el oxígeno carbonilo, que forma un intermedio tetraédrico que colapsa con el procesamiento ácido.[34]
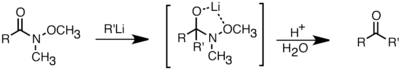
Li add to weinreb

Los reactivos de organolitio también reaccionan con el dióxido de carbono para formar, después del procesamiento, ácidos carboxílicos.[35]
En el caso de sustratos de enona, donde son posibles dos sitios de adición nucleófila (adición 1,2 al carbono carbonilo o adición conjugada 1,4 al carbono β), la mayoría de las especies de organolitio altamente reactivas favorecen la adición 1,2, sin embargo, hay varias formas de impulsar los reactivos de organolitio para que se sometan a la adición conjugada. En primer lugar, dado que es probable que el aducto 1,4 sea la especie más favorable desde el punto de vista termodinámico, la adición de conjugados se puede lograr mediante el equilibrio (isomerización de los dos productos), especialmente cuando el nucleófilo de litio es débil y la adición 1,2 es reversible. En segundo lugar, la adición de ligandos donantes a la reacción forma especies de litio estabilizadas con heteroátomos que favorecen la adición del conjugado 1,4. En un ejemplo, la adición de un bajo nivel de HMPA al disolvente favorece la adición 1,4. En ausencia de un ligando donante, el catión litio está estrechamente coordinado con el átomo de oxígeno; sin embargo, cuando el catión litio es solvatado por HMPA, la coordinación entre el oxígeno del carbonilo y el ion litio se debilita. Por lo general, este método no se puede usar para afectar la regioselectividad de los reactivos de alquil-litio y aril-litio.[36][37]

1,4vs1,2 addition

Los reactivos de organolitio también pueden realizar una adición nucleófila enantioselectiva a carbonilo y sus derivados, a menudo en presencia de ligandos quirales. Esta reactividad se aplica ampliamente en la síntesis industrial de compuestos farmacéuticos. Un ejemplo es la síntesis de Merck y Dupont de Efavirenz, un potente inhibidor de la transcriptasa inversa del VIH. Se agrega acetiluro de litio a una cetona proquiral para producir un producto de alcohol quiral. La estructura del intermedio de reacción activo se determinó mediante estudios de espectroscopia de RMN en estado de solución y cristalografía de rayos X del estado sólido como un tetrámero cúbico 2:2.[38]
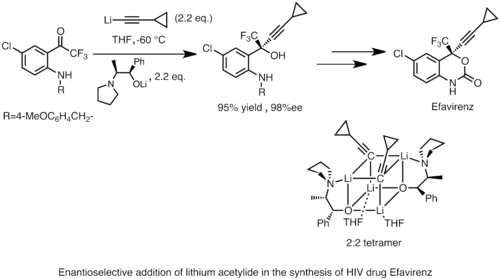
Merck synthesis of Efavirenz

Algunas reacciones generales de compuestos de organolitio son::
- Reacción con sales de ácido carboxílico y de ácido clorhídrico para dar las correspondientes cetona.[39]
- Reacción con ésteres carboxílicos para dar alcoholes terciarios. En el ejemplo adjunto, se demostró que el etilitio era muy eficaz, pero el bromuro de etilmagnesio dio principalmente productos de reducción.[40]

Reacciones tipo SN2
editarLos reactivos de organolitio pueden servir como nucleófilos y llevar a cabo reacciones de tipo SN2 con haluros de alquilo o alilo.[41] Aunque se consideran más reactivos que los reactivos de Grignard en la alquilación, su uso todavía está limitado debido a las reacciones secundarias que compiten, como las reacciones de radicales o el intercambio metal-halógeno. La mayoría de los reactivos de organolitio utilizados en las alquilaciones son más estabilizados, menos básicos y menos agregados, como los reactivos de arilo o alillitio estabilizados con heteroátomos.[12] Se ha demostrado que el HMPA aumenta la velocidad de reacción y el rendimiento del producto, y la reactividad de los reactivos de aril-litio a menudo mejora con la adición de alcóxidos de potasio.[2] Los reactivos de organolitio también pueden realizar ataques nucleofílicos con epóxidos para formar alcoholes.

SN2 inversion with benzyllithium

Reacciones en compuestos de aril-litio
editarLos compuestos aromáticos de litio son adecuados para introducir toda una serie de sustituyentes en el sistema aromático. El alcance de esta reacción comienza con alquilaciones simples, que generalmente reaccionan con un haluro de alquilo. De esta manera también son posibles bromaciones, yodaciones y carboxilaciones. Los ésteres de ácido borónico, como se requiere para el acoplamiento de Suzuki, y los compuestos de organoestaño que se pueden usar para el acoplamiento de Stille también se pueden obtener haciendo reaccionar el organolitio con los cloruros correspondientes.

Como base
editarUn uso común de los compuestos de organolitio simples disponibles comercialmente (como n-BuLi, sec-BuLi, t-BuLi, MeLi o PhLi) es como bases muy fuertes. Los compuestos de organolitio pueden desprotonar casi todos los compuestos que contienen hidrógeno (la metalación o reacción de intercambio Li/H), con la excepción de los alcanos. En principio, una desprotonación puede ir hasta la terminación si el compuesto acído tiene un pKA 2 unidades mayor (ácido 100 veces más fuerte) que el compuesto de litio, aunque en la práctica se requiere una diferencia de pKA más grande para obtener tasas de desprotonación de los ácidos con enlace C-H débilmente ácidos. Como los grupos alquilo son donantes de electrones débiles, la basicidad del compuesto de organolitio aumenta con el número de sustituyentes alquilo sobre el átomo de carbono que soporta la carga.
Los reactivos de organolitio proporcionan una amplia gama de basicidad. El terc-butil-litio, con tres grupos alquilo dadores de electrones débiles, es la base más fuerte disponible comercialmente (pKa = 53). Como resultado, los protones ácidos en −OH, −NH y −SH suelen estar protegidos en presencia de reactivos de organolitio. Algunas bases de litio comúnmente utilizadas son especies de alquillitio como n-butillitio y dialquilamidas de litio (LiNR2). Los reactivos con grupos R voluminosos, como la diisopropilamida de litio (LDA) y la bis(trimetilsilil)amida de litio (LiHMDS), a menudo tienen impedimentos estéricos para la adición nucleófila y, por lo tanto, son más selectivos para la desprotonación. Las dialquilamidas de litio (LiNR2) se utilizan ampliamente en la formación de enolatos y reacciones aldólicas.[7] La reactividad y selectividad de estas bases también están influenciadas por disolventes y otros contraiones.
Desprotonación de iones de organofosfonio
editarLos reactivos de Wittig se utilizan en síntesis orgánica. Se derivan de las sales de fosfonio. Se requiere una base fuerte como el butil-litio o la amida de sodio para la desprotonación:
![{\displaystyle \mathrm {[Ph_{3}P^{+}CH_{2}R]X^{-}+C_{4}H_{9}Li\longrightarrow Ph_{3}P=CHR+LiX+C_{4}H_{10}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/0610431a668579a75779dff43fe098bb59903af0)
Uno de los iluros más simples es el metilentrifenilfosforano (Ph3P=CH2).[42]
Metalación
editarLa metalación con reactivos de organolitio, también conocida como litiación o intercambio de litio-hidrógeno, se logra cuando un reactivo de organolitio, más comúnmente un alquil-litio, extrae un protón y forma una nueva especie de organolitio.
|
|
(4) |

Los reactivos de metalación comunes son los butillitios. El terc-butil-litio y el sec-butil-litio son generalmente más reactivos y tienen mejor selectividad que el n-butil-litio, sin embargo, también son más caros y difíciles de manejar.[8] La metalación es una forma común de preparar reactivos de organolitio versátiles. La posición de la metalación está controlada principalmente por la acidez del enlace C-H. La litiación a menudo ocurre en una posición α para los grupos de extracción de electrones, ya que son buenos para estabilizar la densidad de electrones del anión. Los grupos directores en compuestos aromáticos y heterociclos proporcionan sitios regioselectivos de metalación; La ortometalación dirigida es una clase importante de reacciones de metalación. Las sulfonas metaladas, los grupos acilo y las amidas α-metaladas son intermediarios importantes en la síntesis química. La metalación de alil éter con alquillitio o LDA forma un anión α para el oxígeno y puede proceder a la transposición 2,3-Wittig. La adición de ligandos donantes como TMEDA y HMPA puede aumentar la tasa de metalización y ampliar el alcance del sustrato.[7] Se puede acceder a los reactivos de organolitio quirales mediante metalación asimétrica.[43]

Directed ortho metalation

La ortometalación dirigida es una herramienta importante en la síntesis de compuestos aromáticos sustituidos regioespecíficos. Este enfoque de la litiación y posterior extinción de las especies intermedias de litio con electrófilos suele ser mejor que la sustitución aromática electrófila debido a su alta regioselectividad. Esta reacción procede a través de la desprotonación por reactivos de organolitio en las posiciones α del grupo de metalación directa (DMG) en el anillo aromático. El DMG es a menudo un grupo funcional que contiene un heteroátomo que es básico de Lewis y puede coordinarse con el catión de litio ácido de Lewis. Esto genera un efecto de proximidad inducido por complejos, que dirige la desprotonación en la posición α para formar una especie de arillitio que puede reaccionar con los electrófilos. Algunos de los DMG más efectivos son amidas, carbamatos, sulfonas y sulfonamidas. Son grupos fuertes atractores de electrones que aumentan la acidez de los protones alfa en el anillo aromático. En presencia de dos DMG, la metalación a menudo ocurre orto al grupo director más fuerte, aunque también se observan productos mixtos. Varios heterociclos que contienen protones ácidos también pueden sufrir ortometalación. Sin embargo, para los heterociclos pobres en electrones, generalmente se usan bases de amida de litio como LDA, ya que se ha observado que el alquillitio realiza la adición a los heterociclos pobres en electrones en lugar de la desprotonación. En ciertos complejos de areno-metal de transición, como el ferroceno, el metal de transición atrae la densidad de electrones del areno, lo que hace que los protones aromáticos sean más ácidos y estén listos para la ortometalación.[44]
Superbases
editarLa adición de alcóxido de potasio al alquillitio aumenta en gran medida la basicidad de las especies de organolitio.[45] La "superbase" más común se puede formar mediante la adición de KOtBu al butillitio, a menudo abreviado como reactivos "LiCKOR". Estas "superbases" son reactivos altamente reactivos ya menudo estereoselectivos. En el siguiente ejemplo, la base LiCKOR genera una especie de crotilboronato estereoespecífico a través de la metalación y el posterior intercambio de litio-metaloide.[46]

Superbase

Metalación asimétrica
editarLas especies de organolitio enriquecidas enantio se pueden obtener mediante metalación asimétrica de sustratos proquirales. La inducción asimétrica requiere la presencia de un ligando quiral como (-)-esparteína.[43] La relación enantiomérica de las especies de litio quirales a menudo está influenciada por las diferencias en las tasas de desprotonación. En el siguiente ejemplo, el tratamiento de N-Boc-N-bencilamina con n-butil-litio en presencia de (-)-esparteína proporciona un enantiómero del producto con alto exceso enantiomérico. La transmetalación con cloruro de trimetilestaño produce el enantiómero opuesto.[47]

Asymmetric synthesis with nBuLi and (-)-sparteine
-sparteine.png)
Formación de enolatos
editarLos enolatos de litio se forman a través de la desprotonación de un enlace C-H α con el grupo carbonilo por una especie de organolitio. Los enolatos de litio se utilizan ampliamente como nucleófilos en reacciones de formación de enlaces carbono-carbono, como la condensación aldólica y la alquilación. También son un intermediario importante en la formación de éter silílico de enol.

Sample aldol reaction with lithium enolate

La formación de enolato de litio se puede generalizar como una reacción ácido-base, en la que el protón α relativamente ácido del grupo carbonilo (pK = 20-28 en DMSO) reacciona con la base de organolitio. Generalmente, se utilizan bases fuertes no nucleofílicas, especialmente amidas de litio tales como LDA, LiHMDS y LiTMP. El THF y el DMSO son solventes comunes en las reacciones de enolato de litio.[48]
La estereoquímica y el mecanismo de formación de enolatos han ganado mucho interés en la comunidad química. Muchos factores influyen en el resultado de la estereoquímica del enolato, como los efectos estéricos, el disolvente, los aditivos polares y los tipos de bases de organolitio. Entre los muchos modelos utilizados para explicar y predecir la selectividad en la estereoquímica de los enolatos de litio se encuentra el modelo de Irlanda.[49]
En esta suposición, un LDA monomérico reacciona con el sustrato de carbonilo y forma un estado de transición cíclico de tipo Zimmerman-Traxler. El (E)-enolato se ve favorecido debido a una interacción desfavorable sin-pentano en el estado de transición (Z)-enolato.[48]
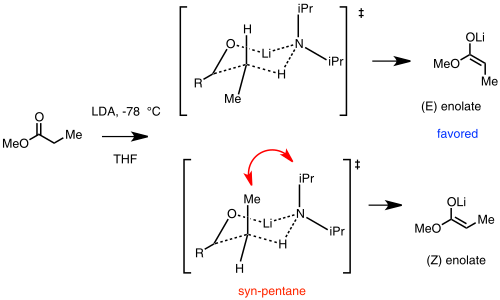
Ireland model for lithium enolate stereoselectivity. In this example, the (E) enolate is favored.

Intercambio halógeno-litio
editarEl intercambio de halógenos por litio es una parte crucial de la ciclización de Parham.[50] En esta reacción, un halogenuro de arilo (comúnmente yoduro o bromuro) se intercambia con un organolitio para formar un areno litiado. Si el areno porta una cadena lateral con un sustituyente electrofílico, el carbanión asociado al litio llevará a cabo un ataque nucleofílico intramolecular para formar una estructura cíclica nueva. Esta reacción es una estrategia útil para la formación de heterociclos.[51] En el ejemplo mostrado, la ciclización de Parham se utiliza en la síntesis de una isoindolinona, a partir de un bromuro de o-metilisocianato arilo. La isoindolinona es precursor de nitronas que son útiles en el estudio de especies reactivas radicalarias de sistemas biológicos.[52]

Transmetalación
editarUna utilidad importante de los reactivos de organolitio es para la preparación de otros compuestos organometálicos, habitualmente por reacción con haluros de metal, como los compuestos de organocinc mediante transmetalación con sales de zinc, como el cloruro de zinc, ZnCl2:[53]

Organozinc reagents from alkyllithium

Especialmente importante en química orgánica de síntesis es la formación de reactivos de organocobre (incluido el reactivo de Gilman) por reacción de alquil-litio, RLi, con yoduro de cobre (I), CuI, o con CuBr. Incluso los reactivos de Grignard se preparan a veces por reacción de alquillitio, RLi, con bromuro de magnesio, MgBr2, en situaciones donde el reactivo de litio (pero no los compuestos de Grignard) puede prepararse fácilmente por reacción de metalación. Los compuestos de Organoestaño, organosilicio, organoboro, organofósforo, y organoazufre se preparan también frecuentemente por reacción de alquil-litio, RLi, con el electrófilo apropiado. Los diorganocupratos de litio se pueden formar haciendo reaccionar especies de alquil litio con haluro de cobre (I). Los organocupratos resultantes son generalmente menos reactivos con los aldehídos y las cetonas que los reactivos de organolitio o los reactivos de Grignard.[54]

1,4 cuprate addition

Otras reacciones
editar- Reacción con oximas para dar las correspondientes aminas.[2]
- Reacción con isonitrilos para dar los correspondientes aldiminas de litio. La posterior hidrólisis efectivamente convierte el compuesto de organolitio en su aldehído.[55]
- Reacción con ciertos epóxidos para dar los correspondientes alquenos.[56]
Véase también
editarEnlaces químicos del carbono con el resto de átomos
editar| CH | He | |||||||||||||||||
| CLi | CBe | CB | CC | CN | CO | CF | Ne | |||||||||||
| CNa | CMg | CAl | CSi | CP | CS | CCl | CAr | |||||||||||
| CK | CCa | CSc | CTi | CV | CCr | CMn | CFe | CCo | CNi | CCu | CZn | CGa | CGe | CAs | CSe | CBr | CKr | |
| CRb | CSr | CY | CZr | CNb | CMo | CTc | CRu | CRh | CPd | CAg | CCd | CIn | CSn | CSb | CTe | CI | CXe | |
| CCs | CBa | CHf | CTa | CW | CRe | COs | CIr | CPt | CAu | CHg | CTl | CPb | CBi | CPo | CAt | Rn | ||
| Fr | CRa | Rf | Db | CSg | Bh | Hs | Mt | Ds | Rg | Cn | Nh | Fl | Mc | Lv | Ts | Og | ||
| ↓ | ||||||||||||||||||
| CLa | CCe | CPr | CNd | CPm | CSm | CEu | CGd | CTb | CDy | CHo | CEr | CTm | CYb | CLu | ||||
| Ac | CTh | CPa | CU | CNp | CPu | CAm | CCm | CBk | CCf | CEs | Fm | Md | No | Lr | ||||
| Química orgánica básica. | Muchos usos en Química. |
| Investigación académica, pero no un amplio uso. |
Enlace desconocido / no evaluado. |
Referencias
editar- ↑ Stent, M. (2002). «Generation of a Highly Basic and Nucleophilic Organolithium; Isopropyllithium». SyntheticPages (195).
- ↑ a b c d e f g h Advanced Organic Chemistry F.A. Carey R.J. Sundberg 2nd Edition ISBN 0-306-41088-5
- ↑ "Organometallics in Organic Synthesis", Schlosser, M., Ed, Wiley: New York, 1994. ISBN 0-471-93637-5
- ↑ Si-Fodil, M. (1998). «Obtention of 2,2-(diethoxy) vinyl lithium and 2-methyl-4-ethoxy butadienyl lithium by arene-catalysed lithiation of the corresponding chloro derivatives. Synthetic applications». Tetrahedron Lett. 39 (49): 8975-8978. doi:10.1016/S0040-4039(98)02031-0.
- ↑ Cohen, T; Bhupathy. M (1989). «Organoalkali compounds by radical anion induced reductive metalation of phenyl thioethers». Acc. Chem. Res. 22 (4): 152-161. doi:10.1021/ar00160a006.
- ↑ a b Snieckus, V (1990). «Directed ortho metalation. Tertiary amide and O-carbamate directors in synthetic strategies for polysubstituted aromatics». Chem. Rev. 90 (6): 879-933. doi:10.1021/cr00104a001.
- ↑ a b c The Preparation of Organolithium Reagents and Intermediates Leroux.F., Schlosser. M., Zohar. E., Marek. I., Wiley, New York. 2004. ISBN 978-0-470-84339-0
- ↑ a b c Organolithium Reagents Reich, H.J. 2002 https://organicchemistrydata.org/hansreich/resources/organolithium/organolithium_data/orgli-primer.pdf
- ↑ Corey, E.J.; Wollenberg, R.H. (1975). «Useful new organometallic reagents for the synthesis of allylic alcohols by nucleophilic vinylation». J. Org. Chem. 40 (15): 2265-2266. doi:10.1021/jo00903a037.
- ↑ a b c d e f g Stey, Thomas; Stalke, Dietmar (2009). «Lead structures in lithium organic chemistry». PATAI'S Chemistry of Functional Groups. John Wiley & Sons, Ltd. ISBN 9780470682531. doi:10.1002/9780470682531.pat0298.
- ↑ Structure Formation Principles and Reactivity of Organolithium Compounds Viktoria H. Gessner, Christian Daschlein, and Carsten Strohmann Chem. Eur. J. 2009, 15, 3320 – 3334 doi 10.1002/chem.200900041
- ↑ a b c d e f Reich, Hans J. (2013). «Role of Organolithium Aggregates and Mixed Aggregates in Organolithium Mechanisms». Chemical Reviews 113 (9): 7130-7178. PMID 23941648. doi:10.1021/cr400187u.
- ↑ Zabicky, Jacob (2009). «Analytical aspects of organolithium compounds». PATAI'S Chemistry of Functional Groups. John Wiley & Sons, Ltd. ISBN 9780470682531. doi:10.1002/9780470682531.pat0304.
- ↑ a b Jemmis, E.D.; Gopakumar, G. (2009). «Theoretical studies in organolithium chemistry». PATAI'S Chemistry of Functional Groups. John Wiley & Sons, Ltd. ISBN 9780470682531. doi:10.1002/9780470682531.pat0297.
- ↑ a b Streiwieser, A. (2009). «Perspectives on Computational Organic Chemistry». J. Org. Chem. 74 (12): 4433-4446. PMC 2728082. PMID 19518150. doi:10.1021/jo900497s.
- ↑ a b Bickelhaupt, F. M. (2006). «Covalency in Highly Polar Bonds. Structure and Bonding of Methylalkalimetal Oligomers (CH3M)n (M = Li−Rb; n = 1, 4)». J. Chem. Theory Comput. 2 (4): 965-980. PMID 26633056. doi:10.1021/ct050333s.
- ↑ Weiss, Erwin (November 1993). «Structures of Organo Alkali Metal Complexes and Related Compounds». Angewandte Chemie International Edition in English (en inglés) 32 (11): 1501-1523. ISSN 0570-0833. doi:10.1002/anie.199315013.
- ↑ Fraenkel, G.; Qiu, Fayang (1996). «Observation of a Partially Delocalized Allylic Lithium and the Dynamics of Its 1,3 Lithium Sigmatropic Shift». J. Am. Chem. Soc. 118 (24): 5828-5829. doi:10.1021/ja960440j.
- ↑ Fraenkel. G (1995). «The carbon-lithium bond in monomeric arllithium: Dynamics of exchange, relaxation and rotation». J. Am. Chem. Soc. 117 (23): 6300-6307. doi:10.1021/ja00128a020.
- ↑ a b Strohmann, C (2009). «Structure Formation Principles and Reactivity of Organolithium Compounds.». Chem. Eur. J. 15 (14): 3320-3334. PMID 19260001. doi:10.1002/chem.200900041.
- ↑ Power, P.P; Hope H. (1983). «Isolation and crystal structures of the halide-free and halide-rich phenyllithium etherate complexes [(PhLi.Et2O)4] and [(PhLi.Et2O)3.LiBr].». JACS 105 (16): 5320-5324. doi:10.1021/ja00354a022.
- ↑ Collum, D. B. (2008). «Solution Structures of Lithium Enolates, Phenolates, Carboxylates, and Alkoxides in the Presence of N,N,N′,N′-Tetramethylethylenediamine: A Prevalence of Cyclic Dimers». J. Org. Chem. 73 (19): 7743-7747. PMC 2636848. PMID 18781812. doi:10.1021/jo801532d.
- ↑ Amanda C. Jones, Aaron W. Sanders, Martin J. Bevan, and Hans J. Reich (2007). «Reactivity of Individual Organolithium Aggregates: A RINMR Study of n-Butyllithium and 2-Methoxy-6-(methoxymethyl)phenyllithium». J. Am. Chem. Soc. (Communication) 129 (12): 3492-3493. doi:10.1021/ja0689334.
- ↑ Elschenbroich, Christoph (2005). Organometallics. Willey-VCH. ISBN 978-3-527-29390-2.
- ↑ Stanetty, P.; Koller, H.; Mihovilovic, M. (1992). «Directed Ortho-Lithiation of Phenylcarbamic Acid 1,l-Dimethylethyl Ester (N-Boc-aniline). Revision and Improvements». J. Org. Chem. 57: 6833-6837. doi:10.1021/jo00051a030.
- ↑ a b c Fananas, Francisco; Sanz, Roberto (2009). «Intramolecular carbolithiation reactions». PATAI'S Chemistry of Functional Groups. John Wiley & Sons, Ltd. ISBN 9780470682531. doi:10.1002/9780470682531.pat0341.
- ↑ Heinz-Dieter Brandt, Wolfgang Nentwig1, Nicola Rooney, Ronald T. LaFlair, Ute U. Wolf, John Duffy, Judit E. Puskas, Gabor Kaszas, Mark Drewitt, Stephan Glander "Rubber, 5. Solution Rubbers" in Ullmann's Encyclopedia of Industrial Chemistry, 2011, Wiley-VCH, Weinheim. doi 10.1002/14356007.o23_o02
- ↑ Baskaran, D.; Müller, A.H. (2010). «Anionic Vinyl Polymerization». Controlled and living polymerizations: From mechanisms to applications. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA. ISBN 9783527629091. doi:10.1002/9783527629091.ch1.
- ↑ Bailey, W.F. (1989). «Preparation and facile cyclization of 5-alkyn-1-yllithiums». Tetrahedron Lett. 30 (30): 3901-3904. doi:10.1016/S0040-4039(00)99279-7.
- ↑ Ashby, E.C.; Noding, S.R. (1979). «The effects of added salts on the stereoselectivity and rate of organometallic compound addition to ketones». J. Org. Chem. 44 (24): 4371-4377. doi:10.1021/jo01338a026.
- ↑ Yamataka, Hiroshi (2009). «Addition of organolithium reagents to double bonds». PATAI'S Chemistry of Functional Groups. John Wiley & Sons, Ltd. ISBN 9780470682531. doi:10.1002/9780470682531.pat0310.
- ↑ name=adamantone>Landa, S. (1967). «Über adamantan und dessen derivate IX. In 2-stellung substituierte derivate». Collection of Czechoslovak Chemical Communications 72 (2): 570-575. doi:10.1135/cccc19670570.
- ↑ Rubottom, G.M.; Kim, C (1983). «Preparation of methyl ketones by the sequential treatment of carboxylic acids with methyllithium and chlorotrimethylsilane». J. Org. Chem. 48 (9): 1550-1552. doi:10.1021/jo00157a038.
- ↑ Zadel, G.; Breitmaier, E. (1992). «A One-Pot Synthesis of Ketones and Aldehydes from Carbon Dioxide and Organolithium Compounds». Angew. Chem. Int. Ed. 31 (8): 1035-1036. doi:10.1002/anie.199210351.
- ↑ Ronald, R.C. (1975). «Methoxymethyl ethers. An activating group for rapid and regioselective metalation». Tetrahedron Lett. 16 (46): 3973-3974. doi:10.1016/S0040-4039(00)91212-7.
- ↑ Hunt, D.A. (1989). «Michael addition of organolithium compounds. A Review». Org. Prep. Proc. Int. 21 (6): 705-749. doi:10.1080/00304948909356219.
- ↑ Reich, H. J.; Sikorski, W. H. (1999). «Regioselectivity of Addition of Organolithium Reagents to Enones: The Role of HMPA». J. Org. Chem. 64 (1): 14-15. PMID 11674078. doi:10.1021/jo981765g.
- ↑ Collum, D.B. (2001). «NMR Spectroscopic Investigations of Mixed Aggregates Underlying Highly Enantioselective 1,2-Additions of Lithium Cyclopropylacetylide to Quinazolinones». J. Am. Chem. Soc. 123 (37): 9135-9143. PMID 11552822. doi:10.1021/ja0105616.
- ↑ Thomas M. Bare and Herbert O. House (1973). "Methyl Ketones from Carboxylic Acids: Cyclohexyl Methyl Ketone". Org. Synth.; Coll. Vol. 5: 775.
- ↑ Posner, Gary H.; Jae Kyoo Lee, Qiang Wang, Sara Peleg, Martin Burke, Henry Brem, Patrick Dolan, Thomas W. Kensler (1998). «Noncalcemic, Antiproliferative, Transcriptionally Active, 24-Fluorinated Hybrid Analogues of the Hormone 1α,25-Dihydroxyvitamin D3. Synthesis and Preliminary Biological Evaluation». Journal of Medicinal Chemistry 41 (16): 3008-3014. doi:10.1021/jm980031t.
- ↑ Sommmer, L.H.; Korte, W. D. (1970). «Stereospecific coupling reactions between organolithium reagents and secondary halides». J. Org. Chem. 35: 22-25. doi:10.1021/jo00826a006.
- ↑ Wittig; Schoellkopf, U. (1960). «Methylenecyclohexane». Organic Syntheses 40: 66. doi:10.15227/orgsyn.040.0066.. Describes Ph3P=CH2.
- ↑ a b Hoppe, Dieter; Christoph, Guido (2009). «Asymmetric deprotonation with alkyllithium– (−)-sparteine». PATAI'S Chemistry of Functional Groups. John Wiley & Sons, Ltd. ISBN 9780470682531. doi:10.1002/9780470682531.pat0313.
- ↑ Clayden, Jonathan (2009). «Directed metallization of aromatic compounds». PATAI'S Chemistry of Functional Groups. John Wiley & Sons, Ltd. ISBN 9780470682531. doi:10.1002/9780470682531.pat0306.
- ↑ Schlosser, M (1988). «Superbases for organic synthesis». Pure Appl. Chem. 60 (11): 1627-1634. doi:10.1351/pac198860111627.
- ↑ Roush, W.R. (1988). «Enantioselective synthesis using diisopropyl tartrate modified (E)- and (Z)-crotylboronates: Reactions with achiral aldehydes». Tetrahedron Lett. 29 (44): 5579-5582. doi:10.1016/S0040-4039(00)80816-3.
- ↑ Park, Y.S. (1996). «(−)-Sparteine-Mediated α-Lithiation of N-Boc-N-(p-methoxyphenyl)benzylamine: Enantioselective Syntheses of (S) and (R) Mono- and Disubstituted N-Boc-benzylamines». J. Am. Chem. Soc. 118 (15): 3757-3758. doi:10.1021/ja9538804.
- ↑ a b Valnot, Jean-Yves; Maddaluno, Jacques (2009). «Aspects of the synthesis, structure and reactivity of lithium enolates». PATAI'S Chemistry of Functional Groups. John Wiley & Sons, Ltd. ISBN 9780470682531. doi:10.1002/9780470682531.pat0345.
- ↑ Ireland. R. E. (1976). «The ester enolate Claisen rearrangement. Stereochemical control through stereoselective enolate formation». J. Am. Chem. Soc. 98 (10): 2868-2877. doi:10.1021/ja00426a033.
- ↑ Parham, W.P.; Bradsher, C.K. (1982). «Aromatic organolithium reagents bearing electrophilic groups. Preparation by halogen-lithium exchange». Acc. Chem. Res. 15: 300-305. doi:10.1021/ar00082a001.
- ↑ Sotomayor, N.; Lete, E. (2003). «Aryl and Heteroaryllithium Compounds by Metal - Halogen Exchange. Synthesis of Carbocyclic and Heterocyclic Systems». Curr. Org. Chem. 7: 275-300. doi:10.2174/1385272033372987.
- ↑ Quin, C. (2009). «Synthesis of a mitochondria-targeted spin trap using a novel Parham-type cyclization». Tetrahedron 65: 8154-8160. doi:10.1016/j.tet.2009.07.081.
- ↑ Reeder, M.R. (2003). «An Improved Method for the Palladium Cross-Coupling Reaction of Oxazol-2-ylzinc Derivatives with Aryl Bromides». Org. Process Res. Dev. 7 (5): 696-699. doi:10.1021/op034059c.
- ↑ Nakamura, E. (1997). «Reaction Pathway of the Conjugate Addition of Lithium Organocuprate Clusters to Acrolein». J. Am. Chem. Soc. 119 (21): 4900-4910. doi:10.1021/ja964209h.
- ↑ G. E. Niznik, W. H. Morrison, III, and H. M. Walborsky (1988). "1-d-Aldehydes from Organometallic Reagents: 2-Methylbutanal-1-d". Org. Synth.; Coll. Vol. 6: 751.
- ↑ David M. Hodgson, Philip G. Humphreys and Matthew J. Fleming (2008). "Organolithiums and Lithium 2,2,6,6-Tetramethylpiperidide in Reductive Alkylation of Epoxides: Synthesis of (E)-Alkenes". Org. Synth. 85: 1-9.
 Datos: Q420460
Datos: Q420460 Multimedia: Organolithium compounds / Q420460
Multimedia: Organolithium compounds / Q420460